Gene level evaluation#
In this notebook we focus on how to train InterScale using the molecular cartography dataset used from Legnini et al., 2023 which we preprocessed in the previous tutorial.
Requirements:
inference results in the anndata object
trained model to load inference results
Import packages#
import scanpy as sc
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
from pathlib import Path
import os
import warnings
# gene program import
import gseapy as gp
import matplotlib as mpl
import pandas as pd
warnings.filterwarnings("ignore")
import interscale
from interscale.config import load_config
from interscale.evaluation import (
gene_loadings,
calculate_gene_ranks,
latent_rank_report,
get_genes_dim,
calculate_dim_importance,
)
interscale.tl.set_full_reproducibility()
import sys
# Find repo root by locating paths.py
BASE_DIR_PROJECT = Path.cwd().resolve().parent.parent
sys.path.insert(0, str(BASE_DIR_PROJECT))
DATA = "legnini"
RESULTS_DIR = Path(f"{BASE_DIR_PROJECT}/results/{DATA}")
Load data and model#
To load the model, check in the previous tutorial 1_model_training.ipynb in the model training for the path where the model was saved to.
adata = sc.read_h5ad(f"{BASE_DIR_PROJECT}/data/{DATA}_trained.h5ad")
cfg = load_config(Path(f"{BASE_DIR_PROJECT}/config_files/{DATA}_example.yaml"))
interscale.model.CombinedModel._setup_anndata(
adata=adata,
prediction_task=cfg.dataset.prediction_task,
layer_key=cfg.dataset.layer_key,
sample_key_list=cfg.dataset.sample_key,
prediction_obs=cfg.dataset.prediction_obs,
view_registry=False,
)
dual_combined_model = interscale.model.CombinedModel.load(
os.path.join(RESULTS_DIR),
adata,
cfg,
local_component=True,
global_component=True,
)
Loading model from /Users/francesca.drummer/Documents/1_Projects/A3-InterScale/interscale/results/legnini/dual_legnini23_regr_node_44_GCN_self-attn-transformer_model.pt
Unclear checkpoint format. Attempting remapping anyway.
State dict remapping applied. Source detected: unknown
Gene rank analysis#
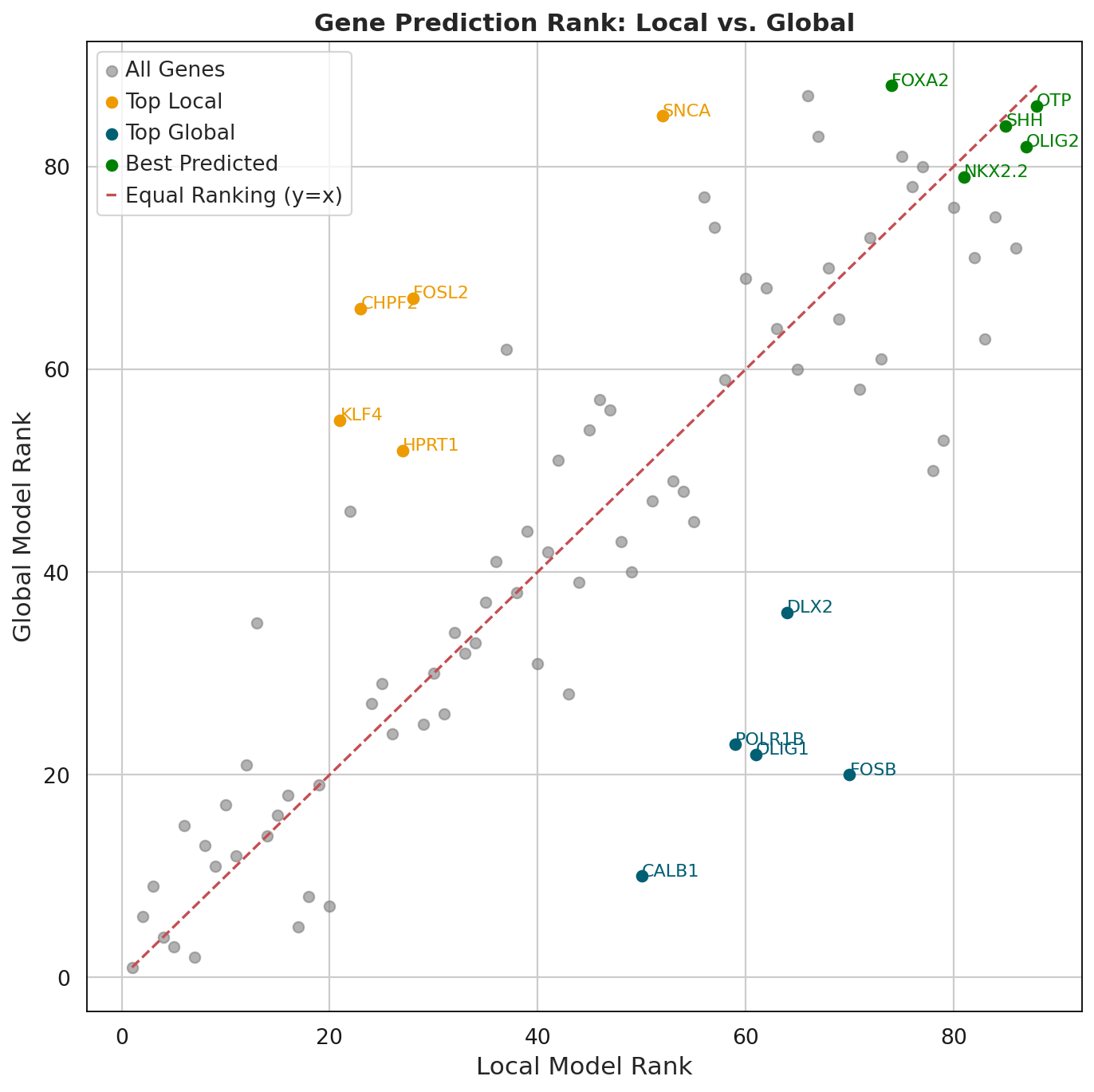
First we check whether the local and global embeddings capture different genes and which genes are better learned by the local vs global component.
For each gene, we computed: Local Rank: Ranking based on local model reconstruction performance XL Global Rank: Ranking based on global model reconstruction performance XG Rank Difference: Local Rank - Global Rank, indicating model preference Average Rank: (Local Rank + Global Rank) / 2, representing overall predictive quality
df = calculate_gene_ranks(adata, layers_local_pred="_y_pred_local", layers_global_pred="_y_pred_global")
interscale.pl.gene_ranks(df, top_n=5)
Gene loadings#
standardize gene loadings meaning = change in gene g (in gene SD units) per 1 SD increase in latent dimension k.
gene_loadings(
adata,
dual_combined_model,
local_latent_key="_global_emb",
global_latent_key="_global_emb",
layer_key="log1p_norm",
)
AnnData object with n_obs × n_vars = 43762 × 88
obs: 'Cell', 'Area', 'x', 'y', 'sample', 'condition', 'organoid', 'obs_names', 'split', '_scvi_sample_key_0', '_scvi_split_key', '_cls_horizontal', '_cls_vertical'
var: 'gene_ids', 'feature_types'
uns: '_scvi_manager_uuid', '_scvi_uuid', 'spatial_neighbors'
obsm: '_attn_matrix', '_global_emb', '_local_emb', 'spatial'
varm: '_local_std_gene_loadings', '_global_std_gene_loadings'
layers: '_y_pred_global', '_y_pred_local', 'log1p_norm', 'norm_ftsqrt', 'raw'
obsp: 'spatial_connectivities', 'spatial_distances'
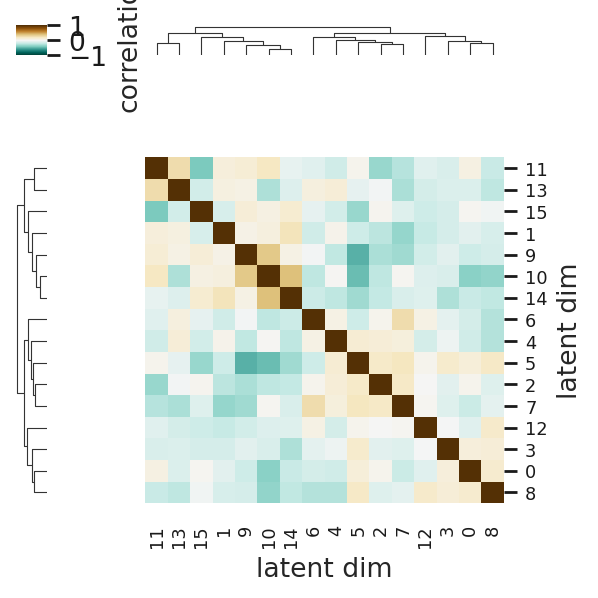
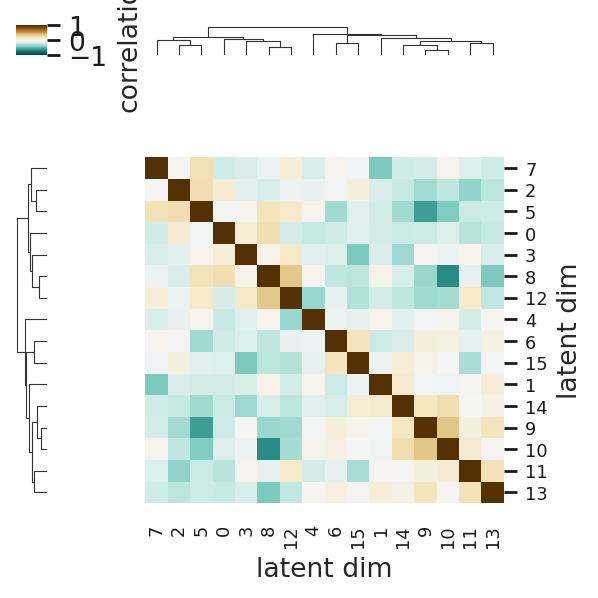
First we check the correlation between each embedding dimension. If the matrix is almost diagonal, we can assume linea independence.
# Check rank:
rep_global = latent_rank_report(adata, z_key="_global_emb")
rep_local = latent_rank_report(adata, z_key="_local_emb")
print(rep_global)
print(rep_local)
{'z_key': '_global_emb', 'n_obs': 43743, 'n_dims': 16, 'rank': 16, 'linearly_independent': True}
{'z_key': '_local_emb', 'n_obs': 43762, 'n_dims': 16, 'rank': 16, 'linearly_independent': True}
interscale.pl.latent_correlation(
adata, z_key="_local_emb", vmax=1.0, cmap="BrBG_r", title="Local Embedding", figsize=(4, 4)
)
interscale.pl.latent_correlation(
adata, z_key="_global_emb", vmax=1.0, cmap="BrBG_r", title="Global Embedding", figsize=(4, 4)
)
<seaborn.matrix.ClusterGrid at 0x3a1640510>
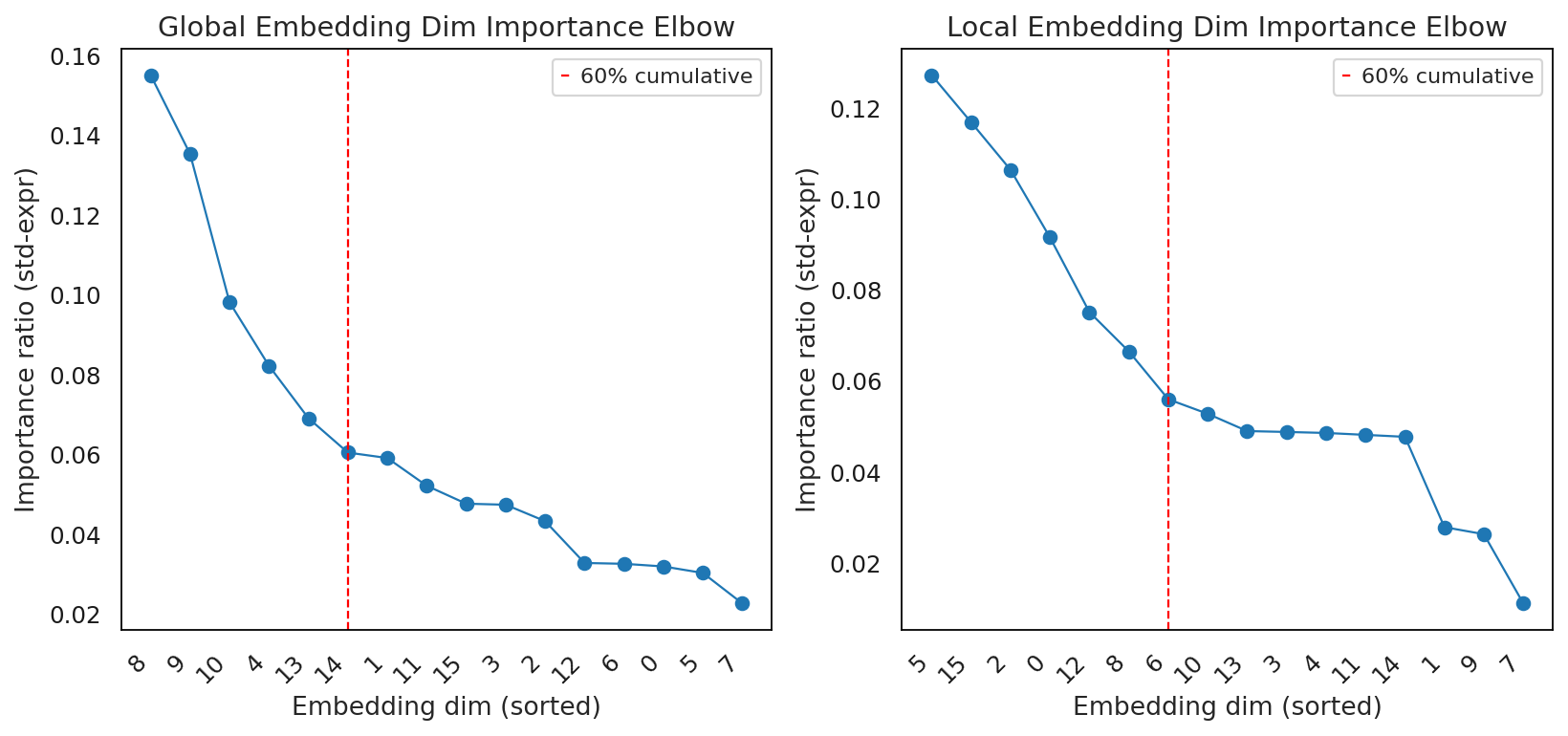
result_global = calculate_dim_importance(
adata,
s_key="_global_std_gene_loadings",
z_key="_global_emb",
mode="full",
use_ratio=True,
cumulative_cutoff=0.60,
spacing=2,
)
result_local = calculate_dim_importance(
adata,
s_key="_local_std_gene_loadings",
z_key="_local_emb",
mode="full",
use_ratio=True,
cumulative_cutoff=0.60,
spacing=2,
)
fig, axes = plt.subplots(1, 2, figsize=(12, 5))
interscale.pl.dim_importance_elbow(result_local, title="Local Embedding Dim Importance Elbow", ax=axes[1])
interscale.pl.dim_importance_elbow(result_global, title="Global Embedding Dim Importance Elbow", ax=axes[0])
60% cumulative variance reached with 7 dimensions:
Embedding dimensions (sorted by importance):
[5, 15, 2, 0, 12, 8, 6]
60% cumulative variance reached with 6 dimensions:
Embedding dimensions (sorted by importance):
[8, 9, 10, 4, 13, 14]
<Axes: title={'center': 'Global Embedding Dim Importance Elbow'}, xlabel='Embedding dim (sorted)', ylabel='Importance ratio (std-expr)'>
Expression Filtering#
Argument |
Effect |
|---|---|
min_frac |
Remove rare genes |
min_sd |
Remove low-variance genes |
Scoring Choices#
Argument |
Meaning |
|---|---|
rank_by=”loading” |
Pure decoder sensitivity |
rank_by=”loading_x_sd” |
Favor highly variable genes |
residualize=True |
Use only unique component of dimension |
Specificity Control#
Argument |
Meaning |
|---|---|
enforce_specificity |
Force genes to be dimension-specific |
specificity_mode=”ratio” |
max/second criterion |
specificity_min |
How strict the specificity must be |
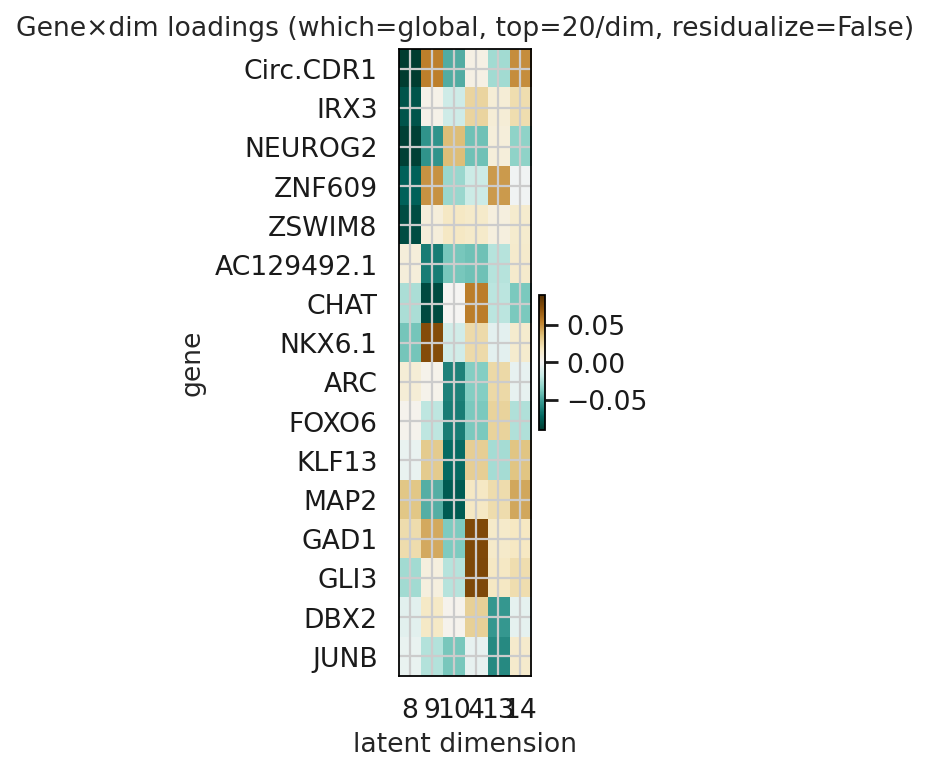
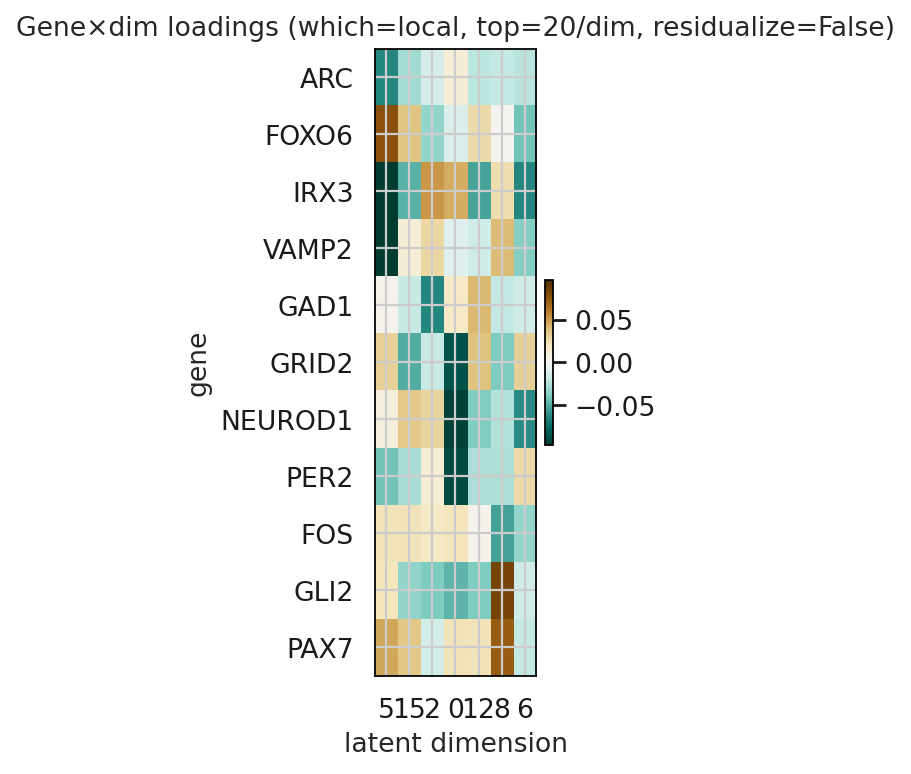
gene_by_dim_heatmap interpretation:
So:
Dark brown → strong positive association with that dimension
Dark green → strong negative association
Near white → little association
Select the local and global dimensions.
global_dim = [8, 9, 10, 4, 13, 14]
local_dim = [5, 15, 2, 0, 12, 8, 6]
df_global, ax = get_genes_dim(
adata,
which="global",
dims=global_dim,
n_top=20,
X_layer="log1p_norm",
min_frac=0.1,
rank_by="loading_x_sd",
enforce_specificity=True,
specificity_min=1.5,
# residualize=True,
plot=True,
figsize=(3.3, 5),
)
df_local, ax = get_genes_dim(
adata,
which="local",
dims=local_dim,
n_top=20,
X_layer="log1p_norm",
min_frac=0.1,
enforce_specificity=True,
rank_by="loading_x_sd",
specificity_min=1.5,
# residualize=True,
plot=True,
figsize=(3.3, 5),
)
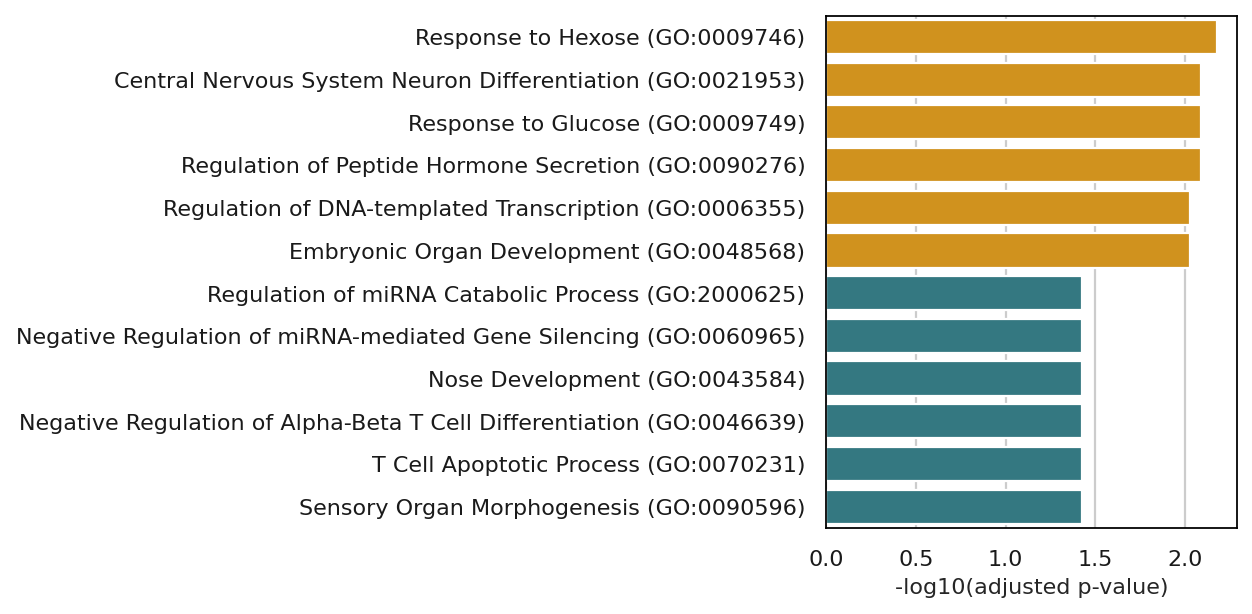
Identify gene programs#
In the next step, we take those top genes associated with the local and global latent dimensions and ask whether they form coherent biological programs.
global_genes = df_global.index
local_genes = df_local.index
shared = global_genes.intersection(local_genes)
unique_local = local_genes.difference(global_genes)
unique_global = global_genes.difference(local_genes)
print("Shared:", shared.tolist())
print("Unique local:", unique_local.tolist())
print("Unique global:", unique_global.tolist())
Shared: ['IRX3', 'ARC', 'FOXO6', 'GAD1']
Unique local: ['FOS', 'GLI2', 'GRID2', 'NEUROD1', 'PAX7', 'PER2', 'VAMP2']
Unique global: ['AC129492.1', 'CHAT', 'Circ.CDR1', 'DBX2', 'GLI3', 'JUNB', 'KLF13', 'MAP2', 'NEUROG2', 'NKX6.1', 'ZNF609', 'ZSWIM8']
enr_global = gp.enrichr(
gene_list=unique_global.tolist(),
gene_sets="GO_Biological_Process_2025",
organism="human", # or "Mouse"
outdir=None,
)
enr_global.results.head()
| Gene_set | Term | Overlap | P-value | Adjusted P-value | Old P-value | Old Adjusted P-value | Odds Ratio | Combined Score | Genes | |
|---|---|---|---|---|---|---|---|---|---|---|
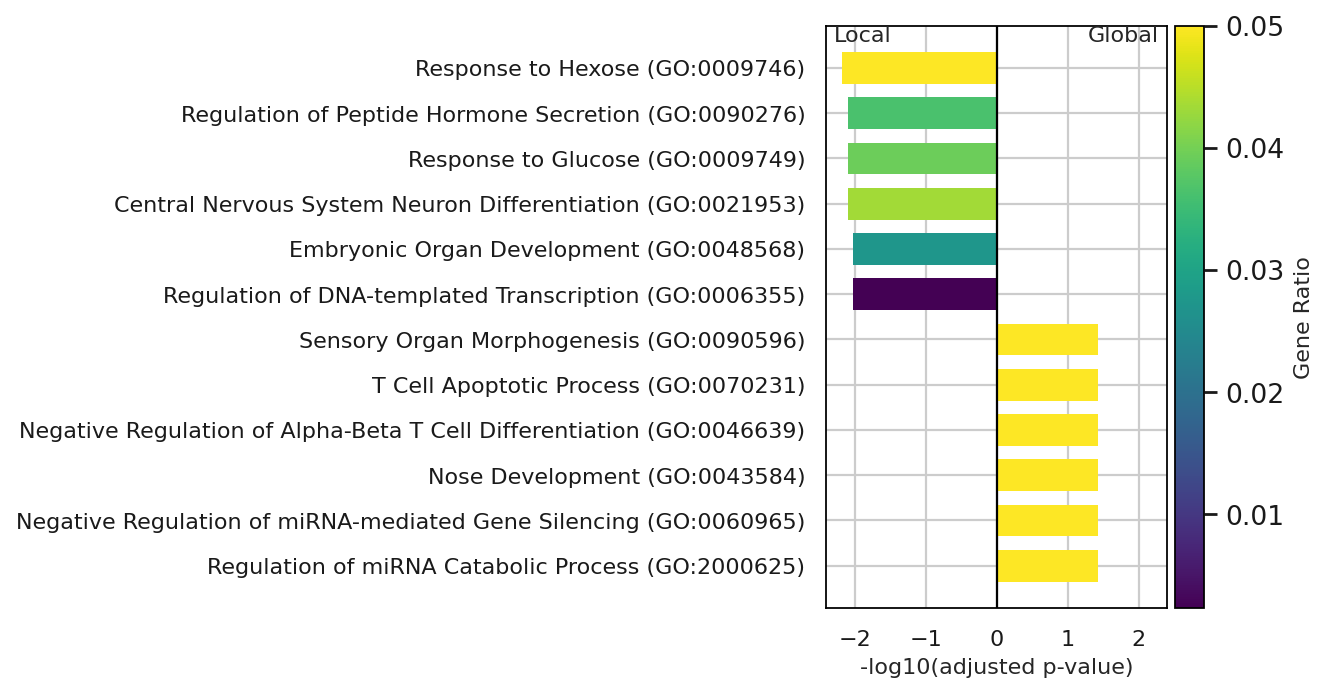
| 0 | GO_Biological_Process_2025 | Regulation of miRNA Catabolic Process (GO:2000... | 1/5 | 0.002997 | 0.037343 | 0 | 0 | 454.181818 | 2638.914119 | ZSWIM8 |
| 1 | GO_Biological_Process_2025 | Negative Regulation of miRNA-mediated Gene Sil... | 1/6 | 0.003595 | 0.037343 | 0 | 0 | 363.327273 | 2044.882815 | ZSWIM8 |
| 2 | GO_Biological_Process_2025 | Nose Development (GO:0043584) | 1/7 | 0.004193 | 0.037343 | 0 | 0 | 302.757576 | 1657.396457 | GLI3 |
| 3 | GO_Biological_Process_2025 | Negative Regulation of Alpha-Beta T Cell Diffe... | 1/8 | 0.004791 | 0.037343 | 0 | 0 | 259.493506 | 1385.975083 | GLI3 |
| 4 | GO_Biological_Process_2025 | T Cell Apoptotic Process (GO:0070231) | 1/8 | 0.004791 | 0.037343 | 0 | 0 | 259.493506 | 1385.975083 | GLI3 |
enr_local = gp.enrichr(
gene_list=unique_local.tolist(),
gene_sets="GO_Biological_Process_2025",
organism="human", # or "Mouse"
outdir=None,
)
enr_local.results.head()
| Gene_set | Term | Overlap | P-value | Adjusted P-value | Old P-value | Old Adjusted P-value | Odds Ratio | Combined Score | Genes | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | GO_Biological_Process_2025 | Response to Hexose (GO:0009746) | 2/25 | 0.000031 | 0.006590 | 0 | 0 | 347.304348 | 3601.323532 | NEUROD1;VAMP2 |
| 1 | GO_Biological_Process_2025 | Central Nervous System Neuron Differentiation ... | 2/46 | 0.000108 | 0.008114 | 0 | 0 | 181.354545 | 1656.578768 | NEUROD1;GRID2 |
| 2 | GO_Biological_Process_2025 | Response to Glucose (GO:0009749) | 2/51 | 0.000133 | 0.008114 | 0 | 0 | 162.808163 | 1453.350008 | NEUROD1;VAMP2 |
| 3 | GO_Biological_Process_2025 | Regulation of Peptide Hormone Secretion (GO:00... | 2/55 | 0.000155 | 0.008114 | 0 | 0 | 150.490566 | 1320.548882 | NEUROD1;PER2 |
| 4 | GO_Biological_Process_2025 | Regulation of DNA-templated Transcription (GO:... | 5/2139 | 0.000243 | 0.009293 | 0 | 0 | 20.921978 | 174.126969 | NEUROD1;PER2;PAX7;FOS;GLI2 |
df_local = enr_local.results
df_global = enr_global.results
df_local["group"] = "Local"
df_global["group"] = "Global"
combined = pd.concat(
[df_local.sort_values("Adjusted P-value").head(6), df_global.sort_values("Adjusted P-value").head(6)]
)
combined["-log10(padj)"] = -np.log10(combined["Adjusted P-value"])
combined = combined.sort_values("-log10(padj)", ascending=False)
palette = {"Global": "#27828E", "Local": "#EE9B00"}
plt.figure(figsize=(8, 4))
sns.barplot(data=combined, y="Term", x="-log10(padj)", hue="group", palette=palette, legend=False)
plt.xlabel("-log10(adjusted p-value)", fontsize=10)
plt.ylabel("", fontsize=10)
# plt.title("GO enrichment: global vs local embeddings", fontsize=10)
plt.xticks(fontsize=10)
plt.yticks(fontsize=10)
# plt.legend(title="", fontsize=10)
plt.tight_layout()
# plt.savefig("figures/gene_programs.png", dpi=300, bbox_inches="tight")
plt.show()
# --- Prepare dataframe ---
plot_df = combined.copy().reset_index(drop=True)
# --- Compute Gene Ratio ---
plot_df["gene_ratio"] = plot_df["Overlap"].str.split("/").apply(lambda x: int(x[0]) / int(x[1]))
# --- Clip extreme values (stabilize color contrast) ---
clip_max = 0.05 # adjust if needed
plot_df["gene_ratio_plot"] = plot_df["gene_ratio"].clip(upper=clip_max)
# --- Signed values for diverging bars ---
plot_df["signed_log10"] = plot_df["-log10(padj)"]
plot_df.loc[plot_df["group"] == "Local", "signed_log10"] *= -1
# --- Sort by absolute significance ---
plot_df = plot_df.sort_values(by="signed_log10", key=lambda s: s.abs(), ascending=True).reset_index(drop=True)
# --- Colormap setup ---
cmap = plt.cm.viridis
norm = mpl.colors.Normalize(vmin=plot_df["gene_ratio_plot"].min(), vmax=plot_df["gene_ratio_plot"].max())
# --- Create figure ---
fig, ax = plt.subplots(figsize=(8, 4.5))
# --- Plot bars ---
ax.barh(
y=plot_df["Term"],
width=plot_df["signed_log10"],
color=cmap(norm(plot_df["gene_ratio_plot"])),
edgecolor="none",
height=0.7,
)
# --- Center line ---
ax.axvline(0, color="black", linewidth=1)
# --- Symmetric x-limits ---
max_val = plot_df["-log10(padj)"].max()
ax.set_xlim(-max_val * 1.1, max_val * 1.1)
# --- Labels ---
ax.set_xlabel("-log10(adjusted p-value)", fontsize=10)
ax.set_ylabel("")
ax.tick_params(axis="x", labelsize=10)
ax.tick_params(axis="y", labelsize=10)
# --- Optional group labels ---
ax.text(-max_val * 1.05, len(plot_df) - 0.5, "Local", ha="left", va="bottom", fontsize=10)
ax.text(max_val * 1.05, len(plot_df) - 0.5, "Global", ha="right", va="bottom", fontsize=10)
# --- Colorbar ---
sm = mpl.cm.ScalarMappable(cmap=cmap, norm=norm)
sm.set_array([])
cbar = fig.colorbar(sm, ax=ax, pad=0.02)
cbar.set_label("Gene Ratio", fontsize=10)
# --- Layout & save ---
plt.tight_layout()
plt.savefig("../../../figures/gene_programs_diverging_gene_ratio.png", dpi=300, bbox_inches="tight")
plt.show()